Une nouvelle méthode pour la recherche de motifs de régulation chez les bactéries en intégrant des données transcriptomiques
Résumé court :
Les séquences génomiques et les données transcriptomiques sont de plus en plus faciles à obtenir. Par contraste, l’intégration de ces sources d’information pour améliorer notre connaissance des réseaux de régulation génétique reste difficile et en grande partie manuelle, même pour des organismes aussi simples que les bactéries. Pour répondre à ce défi, nous avons développé une nouvelle approche dédiée à l’identification des motifs régulateurs dans les séquences d’ADN des promoteurs. Cette méthode fondée sur un modèle statistique original dont les paramètres sont estimés avec un algorithme MCMC transdimensionnel permet un usage simultané des propriétés de composition de l’ADN et de deux types de données transcriptomiques : les positions exactes des sites d’initiation de la transcription et les profils d’expression des gènes à travers les conditions. Pour démontrer sa pertinence, la méthode a été appliquée à un grand jeu de données publique agrégeant les résultats de nombreuses études sur la bactérie Listeria monocytogenes. Les résultats apportent un éclairage global sur les réseaux de régulation de cette bactérie qui est à la fois un pathogène modèle et l’agent responsable d’une des principales anthropozoonoses. Couplée à l’acquisition de données transcriptomiques, la méthode peut être appliquée à n’importe quelle bactérie dans les domaines d’intérêt INRAE car elle ne nécessite pas de disposer d’outils de manipulation génétique.
Contexte et enjeux :
Depuis plus de dix ans l’équipe StatInfOmics de MaIAGE s’intéresse au développement et à l’application de méthodes informatiques et statistiques pour l’exploitation des données de transcriptomique microbienne, accompagnant ainsi le développement rapide des méthodes expérimentales dans ce domaine.
Un des axes de notre travail concerne l’automatisation de l’utilisation des données transcriptomiques pour reconstruire les réseaux de régulation de l’expression génétique via la recherche, dans les séquences d’ADN, de motifs reconnus par des protéines régulatrices. Il s’agit en effet d’une tâche importante mais laborieuse et souvent incrémentale. Tandis que la plupart des algorithmes développés pour automatiser cette tâche abordent le problème sous l’angle de la recherche de motifs expliquant les données transcriptomiques, nous cherchons à tirer parti des données transcriptomiques pour mieux modéliser les propriétés des séquences. L’intérêt de cette stratégie originale est de promettre un continuum entre les méthodes de recherche de motifs avec et sans données transcriptomiques, combinant le meilleur des deux mondes.
Une grande partie de ces travaux ont été menés dans le cadre de projets européens avec des partenaires microbiologistes (EU FP6 Basysbio et FP7 Basynthec coordination P. Noirot, ITN List_Maps, coordination P. Piveteau).
Résultats :
Nous avons publié en 2012 une première méthode implémentant cette stratégie appliquée à la bactérie modèle Bacillus subtilis (Nicolas et al., 2012). Cette méthode visait spécifiquement à identifier les motifs reconnus par les facteurs sigma. Ceux-ci gouvernent la reconnaissance des régions promotrices et sont responsables d’un premier niveau de régulation très important chez certaines bactéries, dont B. subtilis. Pour cela nous avons proposé de résumer l’information des données transcriptomiques sous la forme d’un arbre de classification hiérarchique. Cette méthode a ensuite été appliquée avec succès à la bactérie pathogène Staphylococcus aureus (Mäder et al., 2016).
En 2020, nous avons publié une seconde méthode visant à s’appliquer à l’ensemble des motifs régulateurs (Ibrahim et al., 2020). Pour cela les données transcriptomiques sont prises en compte comme une multitude de covariables synthétisant l’information de co-régulation sous la forme de coordonnées dans des projections sur des axes pertinents (PCA, ICA) ou de position dans des arbres de classification hiérarchique. Un lien de type ‘probit’ est utilisé pour relier ces covariables aux probabilités de présence/absence des motifs. Les paramètres et la dimension du modèle, dont le choix des covariables informatives pour chaque motif, sont ajustés automatiquement pour de nombreux motifs grâce à un algorithme MCMC. La méthode est appliquée à des données publiques disponibles pour la bactérie Listeria monocytogenes.
Perspectives :
Ces méthodes d’identification de motifs régulateurs sont applicables à toutes les bactéries d’intérêt pour INRAE, notamment sur les thèmes Santé Globale et Bioéconomie. L’équipe travaille en particulier sur des bactéries à gram-positif d’intérêt industriel, agro-alimentaire et médical avec l’unité MICALIS (département Microbiologie et chaîne alimentaire) et sur des bactéries pathogènes des poissons d’élevage avec l’unité VIM (département Santé Animale). Le modèle statistique développé pour prendre en compte des covariables de type vecteurs et arbres pourra être transposé à d’autres problématiques d’intégration de données.
Valorisation :
Logiciel libre : https://forgemia.inra.fr/pierre.nicolas/multiple
Références bibliographiques :
P. Nicolas, U. Mäder, E. Dervyn, (47 authors), and P. Noirot. (2012) Condition-Dependent Transcriptome Reveals High-Level Regulatory Architecture in Bacillus subtilis. Science. 335. 1099-1103. (DOI: 10.1126/science.1206848).
U. Mäder, P. Nicolas, (18 authors), and J.M. van Dijl (2016). Staphylococcus aureus Transcriptome Architecture: From Laboratory to Infection-Mimicking Conditions. PLoS Genet. 12. e1005962. (DOI: 10.1371/journal.pgen.1005962).
I. Sultan, V. Fromion, S. Schbath, and P. Nicolas (2020) Statistical modelling of bacterial promoter sequences for regulatory motif discovery with the help of transcriptome data: application to Listeria monocytogenes. J R Soc Interface. 17, 171, 20200600. (DOI: 10.1098/rsif.2020.0600).
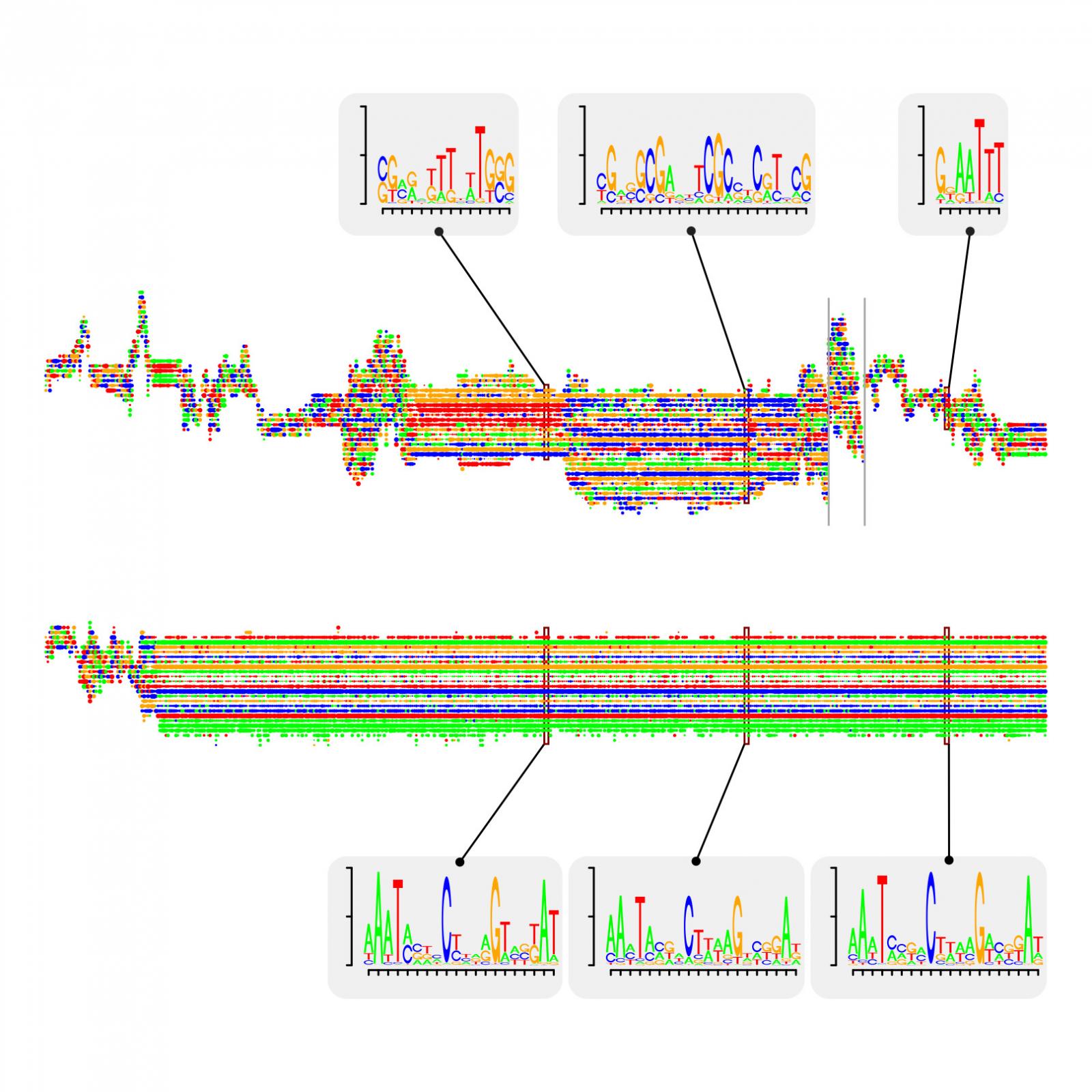
Exploration de l’espace des motifs par l’algorithme MCMC pour l’identification simultanée de nombreux motifs dans les séquences d’ADN des promoteurs. Illustration pour deux motifs. La dimension horizontale décrit la trajectoire d’évolution des motifs au cours des itérations de l’algorithme. La dimension verticale montre la taille et la composition de ces motifs avec un point par position dans le motif et une couleur par type de nucléotide préféré (A, C, G, T). Les inserts représentent les motifs sous forme de logos à deux points de la trajectoire.